Ab Ende Mai 2020 sollen Medizinprodukte in Europa leichter rückverfolgbar und identifizierbar sein. Kürzere Meldefristen für die Marktüberwachung treten in Kraft und die Vorschriften für die Erstellung klinischer Daten werden verschärft. Die Rede ist von MDR, der Medical Device Regulation, die für die Patienten vielversprechend klingt und für ein stärkeres Gefühl der Sicherheit sorgt. Für die Hersteller medizinischer Produkte wirkt sie jedoch eher wie eine bedrohliche Welle, die sich langsam vor ihnen auftürmt und bald auf sie einstürzen könnte, wenn sie nicht schnell eine Lösung finden.
Medical Device Regulation (MDR)
Am 5. Mai 2017 fand die offizielle Bekanntmachung der EU-Medizinprodukte-Verordnung statt. Sie vereint die Richtlinie über Medizinprodukte, Medical Device Directive (MDD), und die Richtlinie über aktiv implantierbare Medizinprodukte, Active Implantable Medical Devices (AIMD). Diese Richtlinien galten ursprünglich unabhängig voneinander, werden durch die Medical Device Regulation künftig jedoch als eine große Verordnung wirken. Das Inkrafttreten folgte 20 Tage nach der Veröffentlichung, am 25. Mai 2017. An diesem Tag begann eine dreijährige Übergangsphase, in der die betroffenen Unternehmen alle Vorbereitungen zu treffen haben, um Konformität mit den Regularien herzustellen.
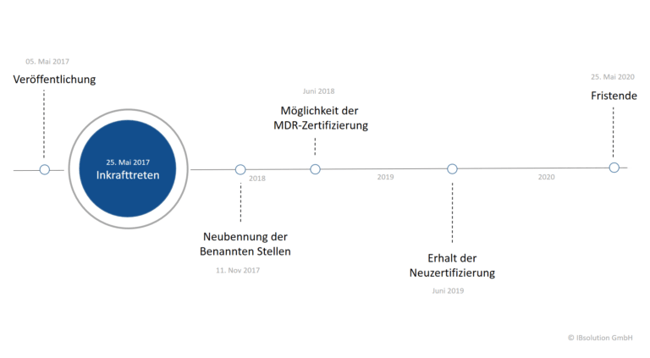
Die eben erwähnte Frist von drei Jahren wird vielerorts kritisiert, sowohl in Fachkreisen als auch vom Bundesverband Medizintechnologie, kurz BVMed. Diese Zeitspanne reiche für die meisten Unternehmen nicht aus. Gründe hierfür sind der langwierige Entwicklungsprozess bei vielen Medizinprodukten, die zahlreichen neuen Anforderungen, die Notwendigkeit zur Einholung einer Freigabe der benannten Stellen und die Vorbereitung auf die neue Europäische Datenbank für Medizinprodukte, die EUDAMED. Hinzu kommt, dass die Herstellerunternehmen in Wirklichkeit keine vollen drei Jahre für die Umsetzung haben. Eine Neubenennung der benannten Stellen, staatlich benannte und überwachte Prüfstellen, war erst nach sechs Monaten, ab dem 26. November 2017, möglich.
Dieses Verfahren nimmt selbst einige Zeit in Anspruch, sodass Anträge der Hersteller auf die MDR-Zertifizierung voraussichtlich erst Ende Juni 2018 möglich sind. Bis sie die Neuzertifizierung erhalten, kann es weitere zwölf Monate dauern. Von den in der Medical Device Regulation versprochenen drei Jahren bleibt also am Ende nur noch ein gutes Jahr übrig.
Wenn Sie die Frist verpassen und Ihre technischen Dokumente am 25. Mai 2020 lückenhaft sind oder Sie kein zugelassenes Zertifikat nachweisen können, haben Sie mit schwerwiegenden Folgen zu kämpfen. Ihnen droht im schlimmsten Fall das Verbot seitens der Behörden, medizinische Produkte in den Verkehr bringen zu dürfen.
Folgen der MDR
Viele der neuen Anforderungen aus den MDR haben starke Auswirkungen auf die IT im Unternehmen. Zu nennen sind:
- Die Angleichung der technischen Dokumentation, die andauernd und in größerem Umfang aktualisiert werden muss.
- Die Umsetzung der UDI-Regelung (Unique Device Identification).
- Die Durchführung der nun genauer geregelten klinischen Bewertung mithilfe von Post-Market-Daten.
- Die Einschätzung potenzieller Sicherheitsrisiken und die damit verbundene Sammlung und Speicherung klinischer Daten.
- Die Neugestaltung der Marktüberwachung mit weiteren Plänen und Berichten.
Unique Device Identification
Zusammen mit den MDR wird auch die UDI, die Unique Device Identification, eingeführt. Ihr Ziel ist es, Medizinprodukte zu identifizieren und zu registrieren und somit rückverfolgbar zu machen. Zukünftig wird es möglich sein, den Lebenszyklus nachzuvollziehen und Produktrückrufe bei auftretenden Problemen einfacher zu gestalten. Zusätzlich sind Produktfälschungen leichter auffindbar und die Sicherheit der Patienten wird im Allgemeinen gesteigert.

Die Unique Device Identification sieht vor, dass die einzelnen Produkte, die von Instrumenten bis hin zu medizinischer Software reichen können, eine individuelle Identifikationsnummer zugeteilt bekommen. Sie muss sich maschinenlesbar und in Klarschrift auf dem Produkt und all seinen Verpackungen befinden. Zwei Teilkomponenten, Device Identifier (DI) und Production Identifier (PI), bilden zusammen die UDI. Der Device Identifier stellt die Produktkennung dar und identifiziert das Medizinprodukt. Der Production Identifier hingegen dient der Herstellungskennung. Er beinhaltet die Seriennummer sowie das Herstellungs- und Verfallsdatum.
Maschinenlesbarkeit wird in diesem System durch einen Strichcode oder eine Datamatrix gewährleistet. Die Erfassung des Strichcodes durch Einscannen vereinfacht Prozesse und vermeidet Fehler, die bei der manuellen Eingabe auftreten können. Dieser Code ist der Schlüssel zur UDID, der Unique Device Identifier Database, mit dem ein öffentlicher Zugang zu spezifischen Informationen des Medizinprodukts gewährleistet wird. In der Datenbank müssen die Hersteller die UDI-DI, das heißt die Produktstammdaten, ihrer Produkte hinterlegen.
Auswirkungen der UDI auf das Stammdatenmanagement
Die UDI-DI eines Medizinprodukts ist gekennzeichnet durch einen einmaligen alphanumerischen oder numerischen Code. Zusammen mit der Produktkennung müssen Produktstammdaten in die UDID eingetragen werden. Zu den relevanten Stammdaten zählen unter anderem der Name und die Risikoklasse des Produkts, der Name des Herstellers, die Art der Kontrolle der Herstellung, die Menge pro Packung, gegebenenfalls wichtige Warnhinweise und die klinische Größe (Länge, Breite, Volumen, Durchmesser).
Falls noch nicht alle Daten zu jedem Produkt des Herstellers vorhanden sind, besteht die Notwendigkeit, sie intern zu erheben. Um einen Überblick über die fehlenden Stammdaten zu erlangen, empfiehlt es sich, einen Report zu erstellen. Er gibt zu erkennen, welche Daten zu welchen Produkten bereits vorhanden sind und welche erst noch erhoben werden müssen. Anschließend findet die Verteilung der Verantwortlichkeiten bezüglich der zu beschaffenden Informationen statt. In welcher Form die Datenerhebung stattfindet, ist jedem Unternehmen selbst überlassen. Es können klassische Excel-Tabellen sein oder andere Softwarelösungen, die auf Stammdatenpflege spezialisiert sind. Der gesamte Vorgang ist, genau wie die nachfolgende Verwaltung der Daten, für die festgelegte Änderungsprozesse gelten, optimalerweise ein transparenter und nachvollziehbarer Workflow mit einer Änderungshistorie.
Bei der Erhebung oder Verwaltung der Produktstammdaten treffen die Unternehmen auf unterschiedlichste Herausforderungen. Zum einen bestehen hohe Anforderungen an die Datenqualität in Bezug auf Vollständigkeit, Korrektheit, systemübergreifende Konsistenz und Aktualität. Ist beispielsweise der in die UDID eingetragene Produktname nicht exakt derselbe wie der Name auf allen Verpackungen, treten Komplikationen auf. Zum anderen wird die Pflege einer Liste mit allen von den Herstellern vergebenen UDIs verpflichtend werden. Sie wird als Teil der technischen Dokumentation aller Medizinprodukte gelten. Zudem kommt es immer wieder zu Veränderungen an den Medizinprodukten, die die Sicherheit oder Zweckbestimmung des Produkts oder die Interpretation der Daten beeinflussen. Tritt mit der Änderung die Gefahr einer Fehlidentifizierung ein oder entstehen Probleme bei der Rückverfolgbarkeit, muss eine neue UDI-DI vergeben werden. Bei Veränderungen an folgenden Aspekten ist das beispielsweise der Fall:
- (Handels-)Name
- Produktmodell oder –version
- Notwendigkeit zur Sterilisation vor Gebrauch
- Warnhinweise oder Kontraindikationen
- Produktmenge in einer Verpackung
- …
Um Voraussetzungen der Datenqualität wie Aktualität und systemübergreifende Konsistenz erfüllen zu können, bieten softwaregestützte Lösungen gegenüber Excel-Listen oder anderen einzelnen Dokumenten praktische Vorteile. Sie dienen als zentraler Speicherort, als sogenannter „Single Point of Truth“. Änderungen und neue Einträge finden nur in dieser Software statt, die somit immer auf dem aktuellsten Stand ist. Auf diese Weise werden Ansammlungen dezentraler Files, lokaler Kopien und zig unterschiedlicher Versionen der Datenbestände vermieden. Zusätzlich lässt sich mithilfe eines Softwaresystems die Zugriffssteuerung regeln. Je nach Aufgabenbereich und Verantwortlichkeiten können den einzelnen Mitarbeitern Zugriffs- und Änderungsrechte zugeteilt werden. Die Funktion einer Änderungshistorie hilft dabei, entstandene Probleme schneller zu identifizieren und gegebenenfalls die ältere Version der Daten wiederherzustellen. Zudem besteht die Möglichkeit, über Workflows Freigabeverfahren bei Stammdatenänderungen zu etablieren.
Fazit
Die zahlreichen Vorschriften bezüglich der Stammdaten in der Unique Device Identification und die näher rückende Frist müssen die Herstellungsunternehmen nicht zwangsläufig in Stress und Panik versetzen. Softwarelösungen im Stammdatenmanagement sind im Markt bereits verfügbar, die in vielen Fällen jedoch noch prozessseitig und inhaltlich an die neue Verordnung angepasst werden müssen.



